
IMGT Education
Présentation des antigènes par MHC-Ia à la surface d'une cellule infectée par un virus à ARN (ex : VIH)
I. Présentation globale du VIH (Virus de l'Immunodéficience Humaine)
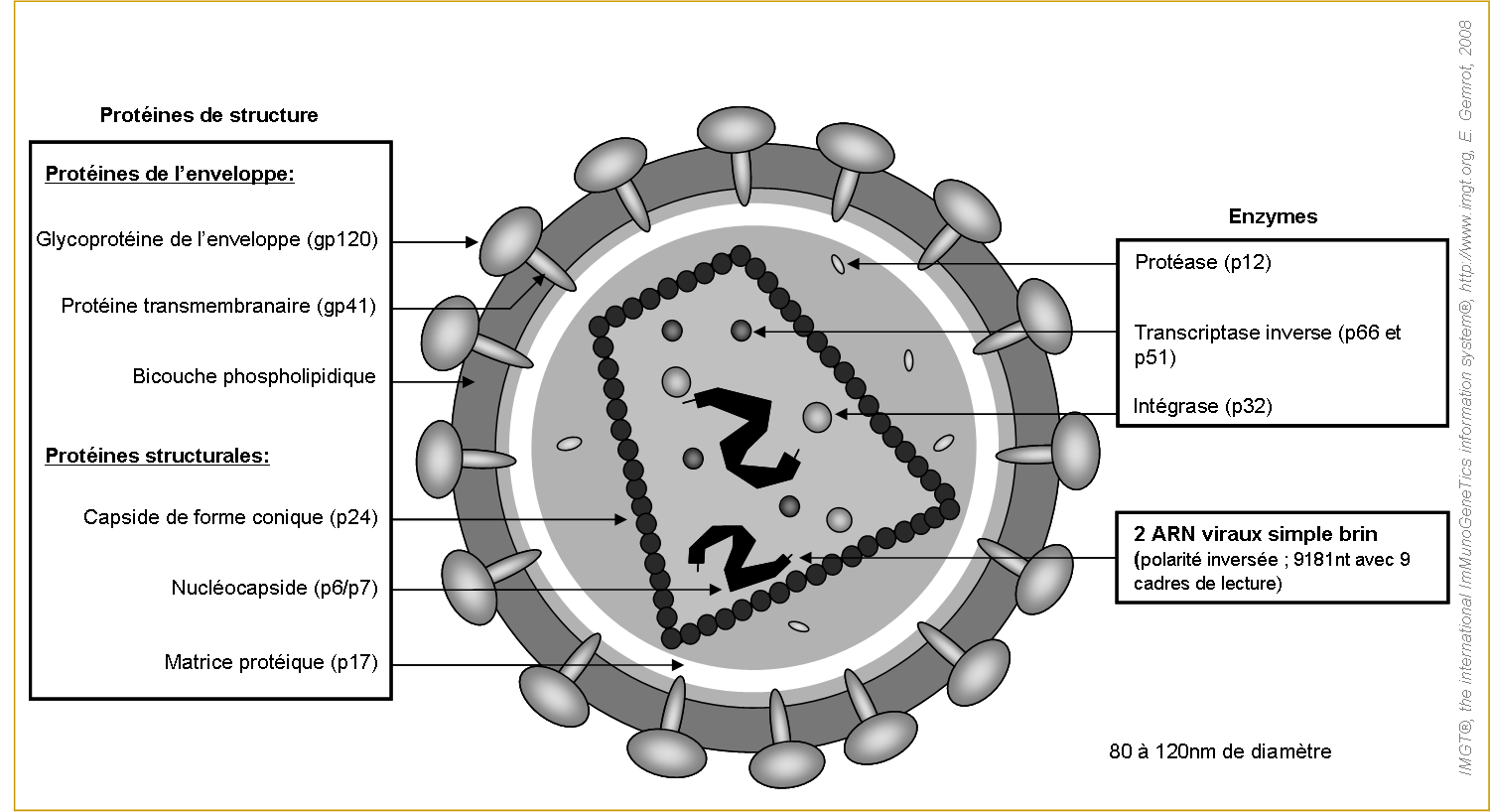 Figure 1. Morphologie du virus VIH.
Figure 1. Morphologie du virus VIH.
La bicouche phospholipidique provient de la membrane de la cellule infectée par le virus.
En plus des enzymes déjà présentes dans le virus, d'autres protéines régulatrices sont exprimées lors du cycle de réplication viral: il s'agit des protéines tat et rev. Lors de la phase tardive du cycle de réplication du virus, des protéines intervenant dans l'assemblage des particules virales et des protéines responsables en partie de la pathogénicité sont exprimées.
Les ARNm viraux codent les protéines structurales (gp120, gp41, p17, p24, p6, p7), les protéines de régulation (p12, p51, p66, p32, tat, rev) ainsi que les protéines nef, vif, vpr et vpu (figure 2).
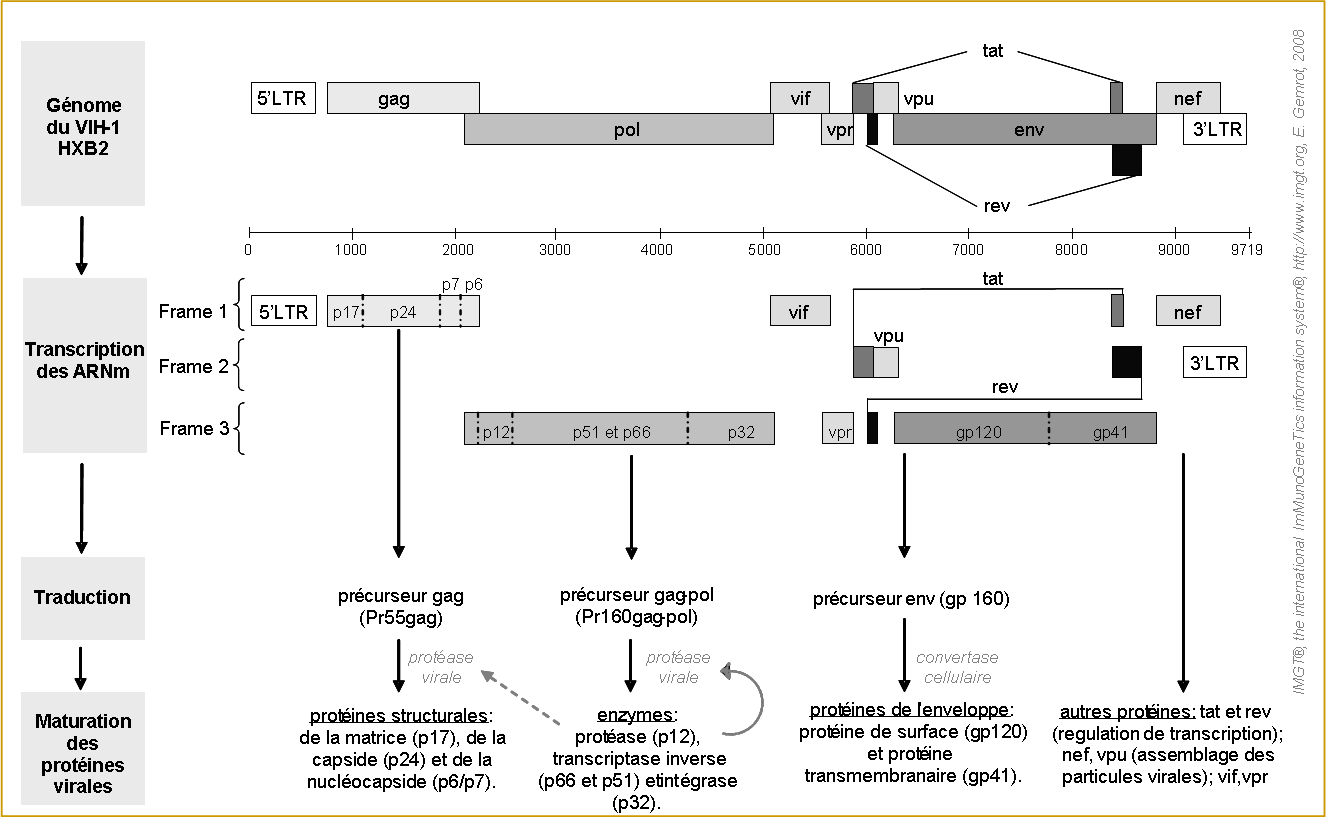 Figure 2. Organisation du génome du virus VIH (Korber et al. 1998 et Nielsen et al. 2005) et expression des gènes.
Figure 2. Organisation du génome du virus VIH (Korber et al. 1998 et Nielsen et al. 2005) et expression des gènes.
L'illustration du génome du VIH-1 HXB2 est réalisée d'après Korber et al. 1998.
La protéase s'autoclive du précurseur gag-pol puis participe à la maturation des protéines de structure et des protéines enzymatiques en clivant les précurseurs protéiques gag et gag-pol.
LTR: long terminal repeat. nef: negative effector. rev: régulator of expression of virion proteins. tat: transactivator of transcription. vif: virion infectivity factor. vpu : viral protein u. vpr : viral protein r.
II. Infection d'une cellule par le virus VIH
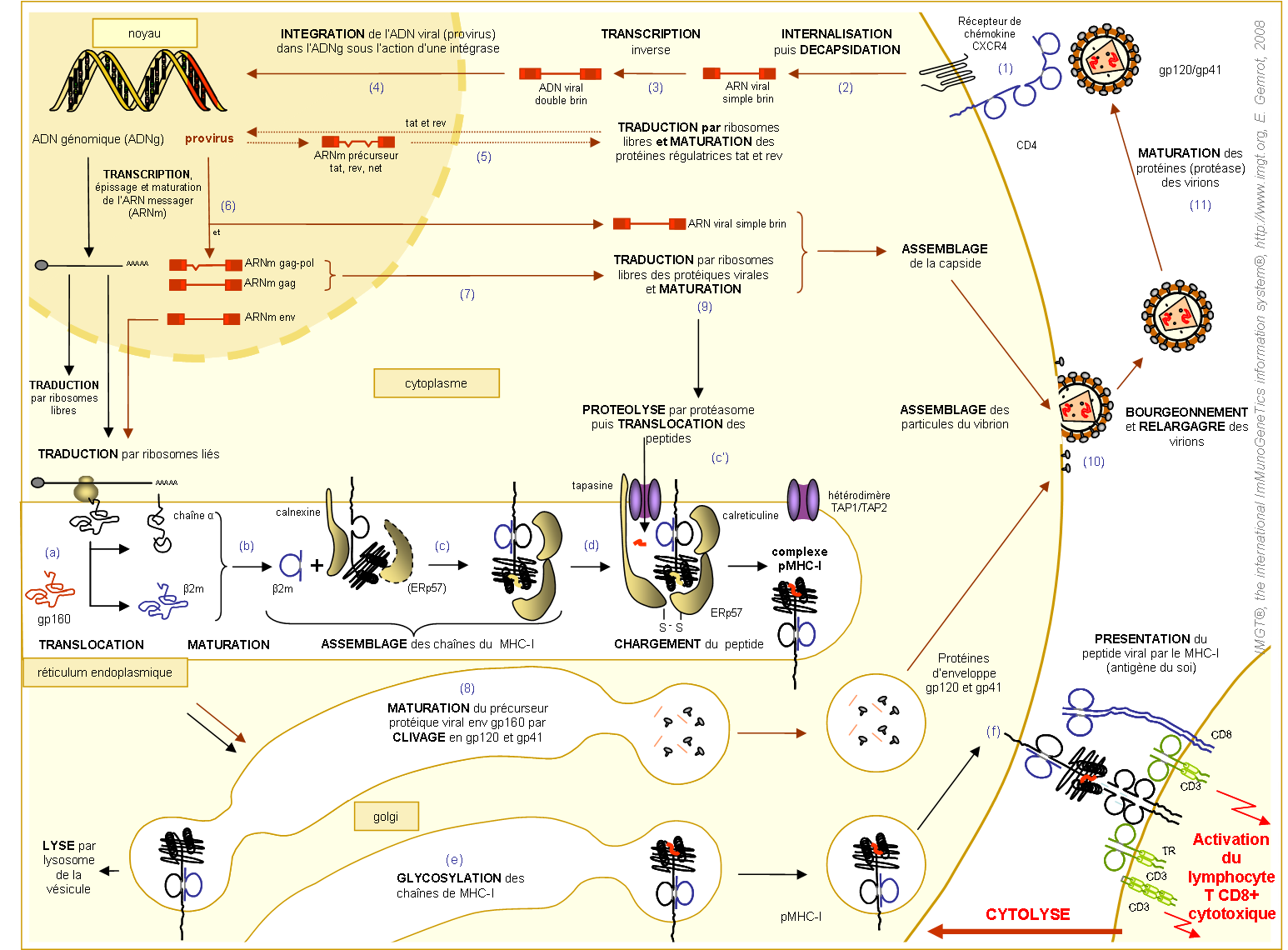 Figure 3. Cycle de réplication du VIH, étapes de l'expression à la surface cellulaire d'un complexe pMHC-Ia et présentation d'un peptide exogène viral par MHC-Ia au récepteur T d'un lymphocyte T CD8+.
Figure 3. Cycle de réplication du VIH, étapes de l'expression à la surface cellulaire d'un complexe pMHC-Ia et présentation d'un peptide exogène viral par MHC-Ia au récepteur T d'un lymphocyte T CD8+.
S'agissant d'un peptide viral (en rouge), le lymphocyte T CD8+ est activé. En résulte la détruction de la cellule infectée par cytolyse. La voie de dégradation des peptides protéiques est indiquée par les lettres « a' » à « c' ». La voie de biosynthèse des MHC-I et de leur chargement en peptides sont indiqués par les lettres « a » à « f ». Le cycle de réplication du VIH est indiqué par les chiffres de « 1 » à « 11 » et détaillé ci-dessous.
Cycle de réplication du virus VIH (figure 3)
Le cycle de réplication des rétrovirus est plus complexe que celui des autres virus à ARN. Ces virus possèdent une enzyme spécifique (la transcriptase inverse) qui transcrit l'ARN viral en ADN.
(1) La pénétration du VIH dans la cellule hôte nécessite le récepteur CD4 et un corécepteur (principalement CCR5 ou CXCR4) (Cocchi et al. 1996, Joshi et al. 2000) (tableau 1). Ces protéines sont essentielles pour la pénétration du virus. Cependant, d'autres molécules de surface de la cellule hôte, tels que les HSPG (héparanes sulfates protéoglycanes) ou encore le LFA-1, peuvent aussi lier le virus.
Tableau 1. Principaux récepteurs du VIH :
| Récepteur | Localisation | Famille | Fonction | Type de récepteur vis-à-vis du VIH | |
|---|---|---|---|---|---|
| CD4 | 12pter-p12 | A la surface des lymphocytes T CD4+. | Récepteur appartenant à la superfamille des immunoglobulines IgSF. | Reconnaissance du MHC-II à la surface des APC par le lymphocyte T et participation à la transduction du signal par le TcR. | Récepteur primaire |
| CCR5 | 3p21 | A la surface des macrophages et des monocytes. | Récepteur de chémokine de type CC (cystéine-cystéine). | Transduction du signal par augmentation de la concentration en Ca2+. | Corécepteur pour la souche HIV-1R5. |
| CXCR4 | 2q21 | A la surface des cellules T. | Récepteur de chémokine de type CXC (cystéine- X-cystéine). | Récepteur primaire pour la souche HIV-2 et corécepteur pour la souche HIV-1X4. | |
Dans un premier temps, la glycoprotéine virale de l'enveloppe (gp120) réalise une liaison avec le récepteur CD4 (figure 4a).
Cette fixation induit un changement de conformation des deux molécules. En résulte, une exposition du site de fixation de la gp 120 (V3 loop) ce qui induit un rapprochement du virus vers la membrane de la cellule hôte et une liaison avec le corécepteur CCR5 ou CXCR4 (figure 4b).
Ces liaisons entrainent des modifications structurales du complexe protéique de l'enveloppe virale. En effet, les protéines gp120 et gp41 du complexe « spike » prennent des conformations qui affaiblissent leur interaction mutuelle (liaisons non covalentes). De ce fait, la glycoprotéine gp120 se dissocie de la protéine transmembranaire gp41. La structure en épingle à cheveux de la gp41 se déplie rendant accessible la région N-terminale des peptides fusion (peptides hydrophobes) qui vont alors s'ancrer dans la membrane de la cellule hôte (figure 4c).
Puis, le domaine HR2 de la gp41 se condense (figure 4d) ce qui entraîne le rapprochement suivit de la fusion des membranes de la particule virale et de la cellule hôte (Nielsen et al. 2005). La capside virale pénètre alors dans le cytoplasme de la cellule cible (Coffin et al. 1996, Gottfredsson et al. 1997 et Tomaras et al. 2001).
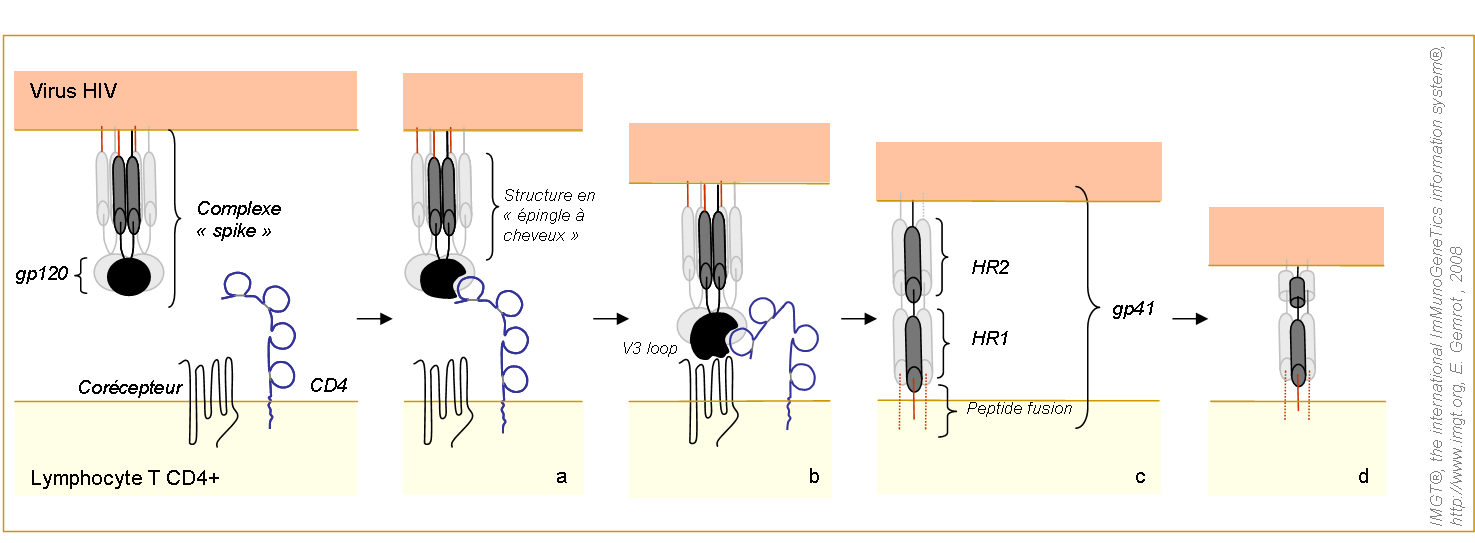 Figure 4. Etapes du processus d'entrée du virus dans une cellule cible.
Figure 4. Etapes du processus d'entrée du virus dans une cellule cible.
Le complexe « spike » est composé d'un trimère de gp41 couplé à un trimère de gp120 de manière non covalente. La protéine gp41 possède une structure dite en épingle à cheveux composée par les régions HR1 et HR2 repliées sur elles mêmes. La protéine gp120 est très variable et possède plusieurs épitopes dont la boucle V3 (V63 loop) qui joue un rôle dans le choix du corécepteur (Nielsen et al. 2005). Les protéines fusion sont symbolisées en rouge.
(2) Des enzymes lytiques de la cellule infectée désassemblent la capside (gp24). De ce fait, les deux ARN viraux et la transcriptase inverse, sont libérés dans le cytoplasme.
(3) Sous l'action de la transcriptase inverse virale et du tRNA3lys de la cellule hôte, l'ARN rétroviral simple brin subit une transcriptase inverse formant ainsi un ADN double brin avec, à ses extrémités, les régions LTR (long terminal repeat) (Joshi et al. 2000 et Coffin et al. 1996).
(4) l'ADN néoformé s'associe avec des protéines cellulaires et virales (vpr et p17) constituant ainsi le complexe de préintégration (Arhel et al. 2007). Ce complexe est ensuite transloqué dans le noyau de la cellule hôte au travers de pores nucléaires (NCP) sous l'action de la vpr. Puis, par le biais de l'intégrase, l'ADN viral est intégré dans le génome de la cellule hôte sous forme de provirus.
l'intégrase virale est constituée de 3 domaines: un domaine N-terminal avec un motif dit « en doigt de zinc », un domaine central catalytique et un domaine C-terminal de liaison à l'ADN « HIV-1 LTR DNA »(Yung et al. 2001 et Zhu et al. 1999).
L'intégration de l'ADN viral est réalisée en deux étapes. Au cours de la première étape, l'enzyme reconnaît les régions LTR « att sites » situées aux deux extrémités du provirus (figure 2). Puis elle les clive spécifiquement sous forme oligomérique. Au cours de la deuxième étape, l'intégrase intègre l'ADN viral dans le génome de la cellule hôte via un mécanisme de transestérification (Coffin et al. 1996, Gottfredsson et al. 1997 et Pommier et al.1999) après avoir clivé, de manière non spécifique, l'ADN de la cellule hôte.
(5) L'expression des gènes viraux peut être divisée en une phase précoce et une phase tardive qui sont respectivement rev-indépendant et rev-dépendant (tableau de la figure 5). Lors de la phase précoce, les ARNm III (codant les protéines tat, rev, nef) sont abondamment transcrits et traduits. L'épissage des ARNm III est réalisé par le complexe d'épissage de la cellule hôte (spliceosome) et traduit dans le cytoplasme de cette dernière. Cela permet d'obtenir les protéines tat et rev nécessaires pour la phase tardive.
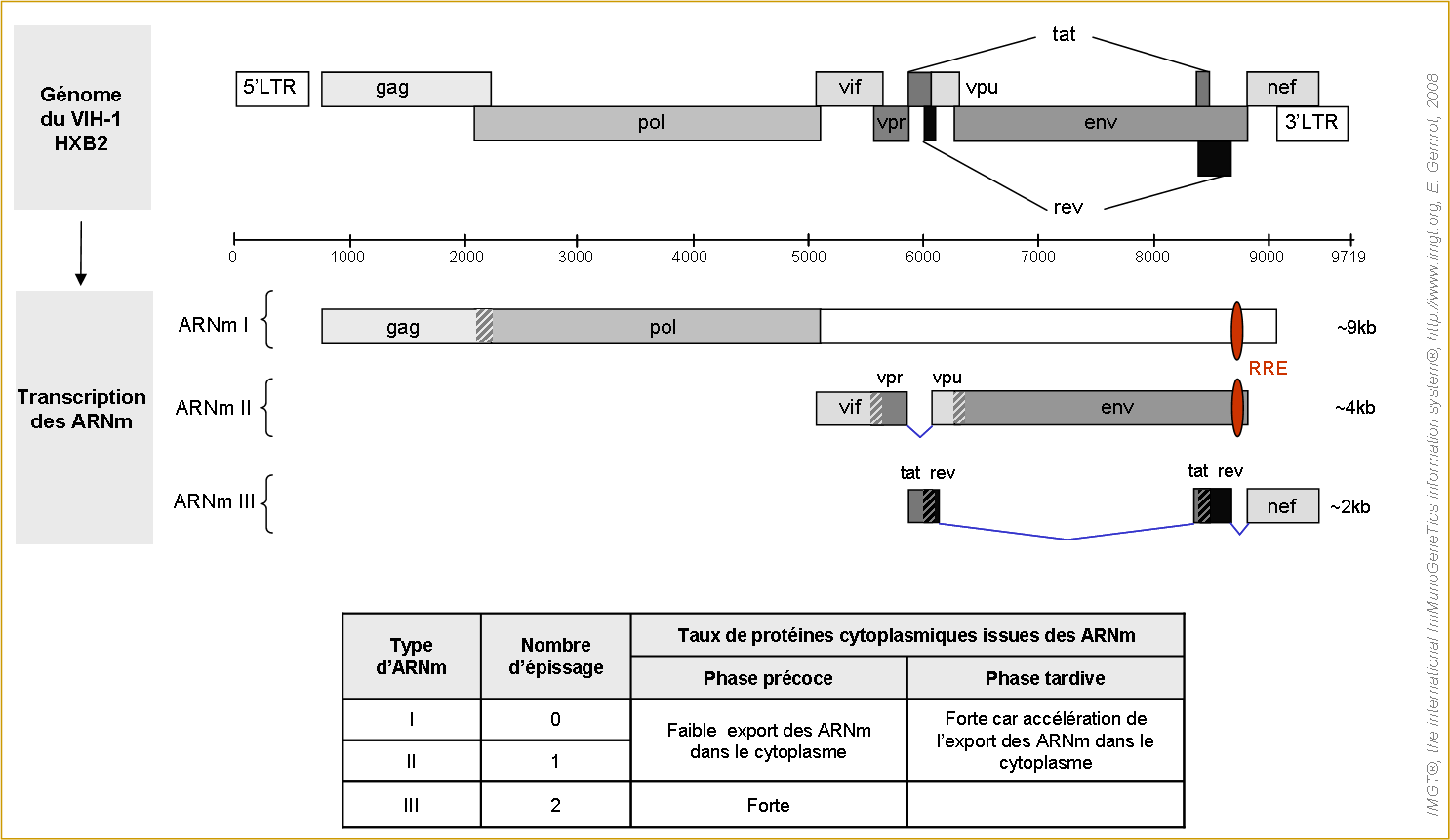 Figure 5. Schéma représentant les différents ARNm et l'action de la protéine rev sur leur expression.
Figure 5. Schéma représentant les différents ARNm et l'action de la protéine rev sur leur expression.
L'illustration du génome du VIH-1 HXB2 est réalisée d'après Korber et al. 1998.
Les zones hachurées correspondent aux séquences communes à deux gènes. Les sites d'épissage sont représentés en bleu. L'ARNm I n'est pas épissé tandis que l'ARNm II et l'ARNm III subissent respectivement un et deux épissages.
Rev contient un motif qui interagit directement avec le palindrome IIB situé au niveau du gène qui code la gp41 (Nielsen et al. 2005). La localisation schématique de RRE (rev response element) est approximative et ne tient pas compte de l'échelle.
(6) Lors de la phase tardive, la transcription des gènes proviraux (ARNm I et ARNm II) dépend de la protéine virale tat et du complexe cellulaire pTEFb. La protéine tat s'associe à la région de trans-activation TAR et au cofacteur de transcription positif cellulaire, P-TEFb (composé de Cyc T1 et CDK9). Le complexe formé stimule le processus d'allongement de la RNA polymérase II cellulaire et, par conséquent, le rendement global de la transcription (Joshi et al. 2000 et Bieniasz et al. 1999) (figure 6).
La régulation de la transcription est également contrôlée par plusieurs facteurs de transcription de la cellule hôte (incluant AP-1, NF-kB, NF-AT, NF-IL-6, CREB, IRF, Sp1, LEF-1/TCF-1α, Ets-1 et USF) (Gottfredsson et al. 1997, Joshi et al. 2000 et Rohr et al. 2003).
|
Figure 6. Complexe d'élongation (Lippincolt et al. 2003). |
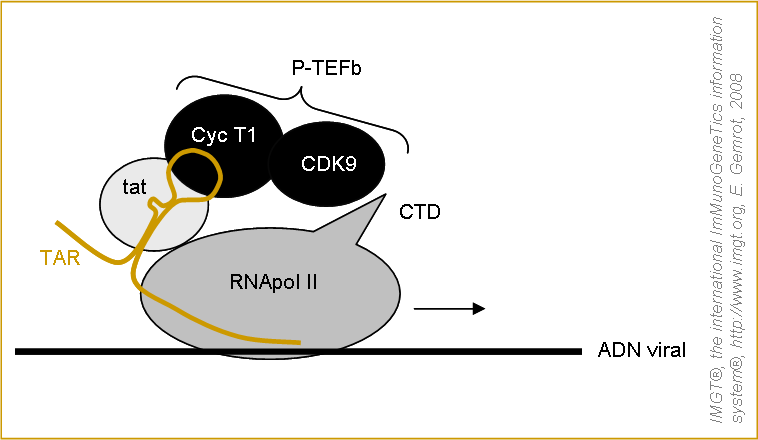 |
La protéine virale rev quant à elle, contrôle le niveau d'expression des différentes protéines virales en régulant le taux d'exportation des ARNm dans le cytoplasme de la cellule infectée (figure 5). Lors de la phase tardive, la protéine rev est, dans un premier temps, importée dans le noyau via l'IMP-β (nuclear import factor inportin). Lorsque la concentration en rev nucléaire atteint un certain seuil, la protéine se lie sur les ARNm I et II au niveau du site RRE (rev response element) sur le gène env. Le complexe ARNm/rev ainsi formé stimule l'exportation des ARNm de type I et II matures vers le cytoplasme où s'effectue la traduction (Nielsen et al. 2005). L'exportation du complexe est régie par le co-facteur cellulaire Cmr1 et l'enzyme Ran-GTP (guanosine triptophate GTPase). Les ARNm ainsi produits codent :
- le précurseur protéique gag (Pr55gag) des protéines structurales de la matrice (p17), de la capside (p24) et de la nucléocapside (p7) ;
- le précurseur protéique gag-pol (Pr160gag-pol) des enzymes protéase (p12), transcriptase inverse (p66/p51) et intégrase (p32) ;
- le précurseur protéique env (gp160) des glycoprotéines de l'enveloppe (gp120) et transmembranaires (gp41) du virus ;
- les protéines vpu (également nommées vpx) intervenant dans l'assemblage viral et les protéines vif et vpr responsables en partie de la pathogénicité.
(7) La traduction des précurseurs gag et gag-pol, codé par l'ARNm I, se fait au niveau du cytoplasme. La maturation des précurseurs gag et gag-pol se fait par l'intermédiaire de la protéase virale p12 après son autoclivage (figure 2).
(8) Parallèlement, la traduction du précurseur env (gp160), codé par l'ARNm II, se fait au niveau des ribosomes liés du réticulum endoplasmique. Lors de la maturation, le précurseur env est clivé, au niveau de l'appareil de golgi, par une convertase cellulaire (furine) en gp120 et gp41 (figure 2).
(9) Les précurseurs des protéines virales cytoplasmiques, le génome viral nouvellement synthétisé et les protéines de l'enveloppe virale se regroupent au niveau de la membrane cellulaire de la cellule hôte pour être assemblés. L'assemblage des particules virales est en partie régi par les protéines vpu, vif et des interactions protéines-protéines (assemblage des protéines de la capside, de la matrice et de la nucléocapside). La protéine vpu interviendrait dans l'adressage des protéines de l'enveloppe tandis que la protéine vif aurait une influence sur les étapes finales d'assemblage des virions (Coffin et al. 1996, Joshi et al. 2000). En effet, cette protéine réalise un contrôle temporel des précurseurs gag, empêchant leur maturation prématurée et par là même garantissant la disponibilité des polypeptides de la capside, de la matrice et de la nucléocapside lors de l'assemblage de composants viraux (Coffin et al. 1996, D'aloja et al. 1998 et Miller et al. 2000). Une fois les précurseurs clivés, les protéines p17 s'assemblent pour former la matrice de l'enveloppe virale, les protéines p24 se condensent pour former une capside conique entourant les ARN viraux eux-même enveloppés par les protéines p6 et p7.
(10) S'en suit le bourgeonnement. Les glycoprotéines membranaires s'incorporent dans la membrane plasmique de la cellule hôte et ultérieurement dans le vibrion naissant.
(11) La nouvelle particule virale est relâchée en dehors de la cellule et subit la fin de sa maturation avant d'entamer un nouveau cycle de réplication (Coffin et al. 1996).
III. Facteurs de virulence du VIH
- multiplication rapide (génome court, ARN dupliqué, utilisation de la machinerie de la cellule hôte) ;
- nombreuses souches recensées existantes ;
- recombinaison de souches différentes (création possible d'un virus hybride si une même cellule est infectée par différentes souches) ;
- variabilité génétique (épitopes multiples du gp120 en particulier dans la boucle V3) ;
- capacité à muter rapidement (due à la la transcriptase inverse) permettant au virus d'être continuellement en avance sur la réponse des cellules du système immunitaire ;
- sélection naturelle des souches virales les plus virulentes et résistantes au système immunitaire (faible variabilité au niveau des gènes gag et pol) ;
- plusieurs cibles possibles : principalement les cellules lymphocytes T CD4+, les natural killers, les cellules dendritiques et les macrophages mais aussi les cellules de la paroi intestinale, les astrocytes et les cellules du système nerveux central ;
- les cellules peuvent être des cellules réservoirs quand le virus est en état de dormance ;
- virus véhiculé par le sang et par la lymphe ;
- maladie longue qui détruit progressivement le système immunitaire ;
- possession de protéines à haut pouvoir infectieux.
Tableau 2. Fonction des principales protéines virales du VIH :
| Protéine virale | Localisation de l'action au niveau de la cellule hôte | Molécules cibles | Pouvoir infectieux | Fonction sur cellule | Fonction sur virus |
|---|---|---|---|---|---|
| tat | Noyau | MHC-II | Camouflage du virus | Empêche la transcription de MHC-IIa, de la chaîne Li, de HLA-DMA et HLA-DMB, en se fixant sur le facteur de transcription P-TEFb. | Permet la transcription des gènes viraux [a]. |
| nef | Réticulum endoplasmique et golgi | Non dégradation de la chaîne Li par perturbation du transport et augmentation de la concentration en protéines de MHC-II immatures. | Favorise l'export des ARNm de type I et II dans le cytoplasme [b]. | ||
| MHC-I | Empêche l'expression de MHC-I à la surface de la cellule infectée par dégradation des MHC-I au niveau des lysosomes. | ||||
| vif | Cytoplasme | APOBEC3G | Non dégradation du virus | Inhibe la fonction de l'enzyme APOBEC3G (cytidine désaminase) par formation d'un complexe empêchant ainsi l'encapsidation de l'ADN viral [c]. | Contrôle temporel de la maturation du précurseur gag [d]. |
| vpu ou vpx | Réticulum endoplasmique et golgi | MHC-I | Camouflage du virus | Empêche l'expression de MHC-I à la surface de la cellule infectée par dégradation des MHC-I au niveau des lysosomes. | Permet l'assemblage des protéines virales et contrôle temporel de l'infection par rapport à la phase du cycle de la cellule infectée [e]. |
(a) Coffin et al. 1996 et Bieniasz et al. 1999. (b) Nielsen et al. 2005. (c) Lecossier et al. 2003 et Sheehy et al. 2002. (d) Coffin et al. 1996, D'aloja et al. 1998 et Miller et al. 2000. (e) Coffin et al. 1996, Joshi et al. 2000 et Gottfredsson et al. 1997.
Il est à noter que les mécanismes d'action de pathogénicité des protéines virales sont encore à l'étude. Par ailleurs, il semblerait que plusieurs stratégies soient utilisées par le virus afin d'enrailler la présentation des peptides virales aux récepteurs T des lymphocytes via les MHC-I et MHC-II. Ainsi on peut citer:
- l'inhibition de la transcription ;
- l'inhibition de la maturation des CMH, d'où destruction des récepteurs dans les lysosomes ;
- l'inhibition du transport des CMH à la surface.
IV. Exemples de peptides VIH présentés par le CMH
Tableau 3. Exemples de peptides VIH présentés par le CMH:
| Protéines | Entrées ID IMGT/3Dstructure-DB | Gènes et allèles IMGT | Description | Ligands |
|---|---|---|---|---|
| Précurseur gag-pol | 2bvq | HLA-B*5703 | MHC-I-ALPHA_B2M | gag-pol polyprotein, P12499, peptide 162-172 |
| Précurseur gag peptide | 1t1w, 1t1y, 1t1z, 1t20, 1t21, 1t22, 1s8d, 2c7u | HLA-A*0201 | MHC-I-ALPHA_B2M | gag |
| 1a1m | HLA-B*5301 | MHC-I-ALPHA_B2M | gag p16, P18095, peptide 182-190 | |
| 2bvo | HLA-B*5703 | MHC-I-ALPHA_B2M | gag, Q70A02, peptide 48-58 | |
| 2bvp | HLA-B*5703 | MHC-I-ALPHA_B2M | gag, Q8URG0, peptide 19-27 | |
| p17 | 1agb | HLA-B*0801 | MHC-I-ALPHA_B2M | gag p17, P03349, peptide 23-30, K3>R |
| 1agc | HLA-B*0801 | MHC-I-ALPHA_B2M | gag p17, P03349, peptide 23-30, K7>Q | |
| 1agd | HLA-B*0801 | MHC-I-ALPHA_B2M | gag p17, P03349, peptide 23-30 | |
| 1age | HLA-B*0801 | MHC-I-ALPHA_B2M | gag p17, P03349, peptide 23-30, K7>R | |
| 1agf | HLA-B*0801 | MHC-I-ALPHA_B2M | gag p17, P03349, peptide 23-30, K5>R [HIV-1] | |
| 2v2w, 2v2x | HLA-A*0201 | MHC-I-ALPHA_B2M | HIV-1 Gag p17 epitope-related peptide | |
| gp120 | 1hhg | HLA-A*0201 | MHC-I-ALPHA_B2M | envelope gp120, P03375, peptide 192-200 |
| rev | 1hhj | HLA-A*0201 | MHC-I-ALPHA_B2M | pol reverse transcriptase, P03366, peptide 476-484 |
| 1i1f | HLA-A*0201 | MHC-I-ALPHA_B2M | pol reverse transcriptase, P03366, peptide 476-484 | |
| 1i1y | HLA-A*0201 | MHC-I-ALPHA_B2M | pol reverse transcriptase, P03366, peptide 476-484 | |
| 1p7q | HLA-A*0201 | MHC-I-ALPHA_B2M | pol reverse transcriptase, P03366, peptide 476-484 | |
| 1q94 | HLA-A*1101 | MHC-I-ALPHA_B2M | pol reverse transcriptase, P03366, peptide 325-333 | |
| 1akj | HLA-A*0201 | MHC-I-ALPHA_B2M | pol reverse transcriptase, P03366, peptide 476-484 | |
| tax | 1duy | HLA-A*0201 | MHC-I-ALPHA_B2M | tax, Q82235, peptide 12-19 |
| 1duz | HLA-A*0201 | MHC-I-ALPHA_B2M | tax, Q82235, peptide 11-19 | |
| nef | 1a1n | HLA-B*3501 | MHC-I-ALPHA_B2M | nef, P03407, peptide 78-85 |
| 1qvo | HLA-A*1101 | MHC-I-ALPHA_B2M | nef, P03407, peptide 77-86 |
V. Bibliographie
Arhel, N.J., Souquere-Besse, S., Munier, S., Souque, P., Guadagnini, S., Rutherford, S., Prevost, M.C., Allen, T.D., Charneau, P., HIV-1 DNA Flap formation promotes uncoating of the pre-integration complex at the nuclear pore, EMBO J., 26, 3025-3037 (2007).
Bieniasz, P.D., Grdina, T.A., Bogerd, H.P., Cullen, B.R., Recruitment of cyclin T1/P-TEFb to an HIV type 1 long terminal repeat promoter proximal RNA target is both necessary and sufficient for full activation of transcription, Proc. Natl. Acad. Sci. USA Biochemistry, 96, 7791-7796 (1999).
Cocchi, F. and al., The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection, Nature Med., 2, 1244-1247 (1996).
Coffin, J.M., Retroviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM., editor, Fields Virology, Third. Philadelphia: Lippincott - Raven Publichers, 1767-1830 (1996).
D'aloja, P., Olivetta, E., Bona, R., Nappi, F., Pedacchia, D., Pugliese, K., Ferrari, G., Verani, P., Federico, M., gag, vif, and nef genes contribute to the homologous viral interference induced by a nonproducer human immunodeficiency virus type 1 (HIV-1) variant: Identification of novel HIV-1-inhibiting viral protein mutants, J. Virol., 72, 4308-4319 (1998).
Gottfredsson, M., Bohjanen, P.R., Human immunodeficiency virus type 1 as a target for gene therapy, Frontiers in Bioscience, 2, 619-634 (1997).
Joshi, S., Lamothe, B., Current developments and future prospects for HIV gene therapy using interfering RNA-based strategies, Frontiers in Bioscience, 5, 527-555 (2000).
Korber, and al., Issu du livre Human retroviruses and AIDS, Numbering positions in HIV relative to HXB2CG, p102 (1998).
Lecossier, D., Bouchonnet, F., Clavel, F., Hance, A.J., Hypermutation of HIV-1 DNA in the absence of the Vif protein, Science, 300, 1112 (2003).
Miller, R.J., Cairns, J.S., Bridges, S., Sarver, N., Human immunodeficiency virus and AIDS: Insights from animal Lentiviruses, J. Virol., 74, 7187-7195 (2000).
Nielsen, M.H, Pedersen, F.S, Kjems, J., Molecular strategies to inhibit HIV-1 replication, Retrovirology, 2, 10 (2005).
Pommier, Y., Neamati, N., Inhibitors of human immunodeficiency virus integrase, Adv. Virus Res., 52, 427-58 (1999).
Rohr, O., Marban, C., Aunis, D., Schaeffer, E., Regulation of HIV-1 gene transcription: from lymphocytes to microglial cells, Journal of Leukocyte Biologi, 74, 736-749 (2003).
Sheehy, A.M., Gaddis, N.C., Choi, J.D., Malim, M.H., Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein, Nature, 418, 646-650 (2002).
Tomaras, G.D., Greenberg, M.L., Mechanisms for HIV-1 entry: Current strategies to interfere with this step, Current Infectious Disease Reports, 3, 93-99 (2001).
Yung, E., Sorin, M., Pal, A., Craig, E., Morozov, A., Delattre, O., Kappes, J., Ott, D., Kalpana, G.V., Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1, Nature Med., 7, 920-926 (2001).
Zhu, K., Cordeiro, M.L., Atienza, J., Robinson, W.E., Chow, S.A., Irreversible inhibition of human immunodefiency virus type 1 integrase by dicaffeoylquinic acids, J. Virol., 73, 3309-3316 (1999).
Editor: Elodie Gemrot
IMGT Home page |
IMGT Repertoire (IG and TR) |
IMGT Repertoire (MH) |
IMGT Repertoire (RPI) |
IMGT Index |
IMGT Scientific chart |
IMGT Education |
IMGT Latest news ![]()



© Copyright 1995-2026 IMGT®, the international ImMunoGeneTics information system® | Terms of use | About us | Contact us | Citing IMGT